6. Case Study 5 – a complex 'grey' case
A boy (III.2), who had been previously well, presented at the age of 18 months with poor feeding, irritability, and vomiting. A blood ammonia was 164 µmol/L and urine orotic acid increased at 1700 µmol/L.
He was commenced on treatment with sodium benzoate and arginine and made a good recovery.
A previously unreported change is found in the OTC gene in the boy and his mother (II.5).
In order to confirm pathogenicity, it would be possible to measure OTC enzyme activity in liver tissue but this invasive investigation is difficult to justify. The increased orotic acid suggests that the hyperammonaemia was probably due to OTC deficiency and determining whether the mutation affects an evolutionarily conserved part of the gene may be helpful.
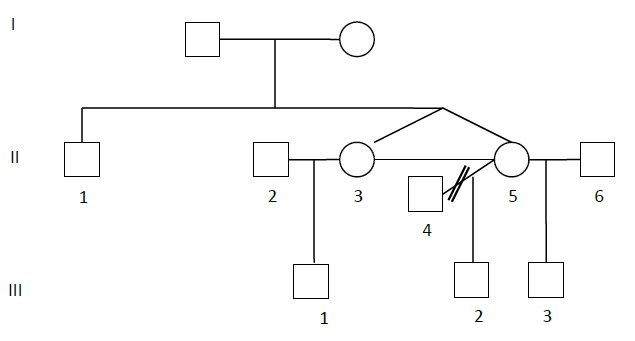
Comment: The horizontal line joining the sisters on the pedigree indicates that they are identical.
On family screening Patient II.1 (see pedigree above) is found to be hemizygous. However at the age of 22 years he is reported to have always been asymptomatic.
Patients with mild OTC deficiency with significant residual enzyme activity may present for the first time at any age. Presumably this man has never been subjected to a catabolic stress or protein load that resulted in hyperammonaemia. There is a 50% chance that he will have inherited the mutation. Since his half-brother did not present in the newborn period careful monitoring of blood ammonia in the newborn period is probably required rather than immediate treatment with medication.
Blood ammonia at 7 days of age was 125 µmol/L with a plasma glutamine of 1153 µmol/L and a plasma citrulline of 4 µmol/L. It is difficult to come to any definite conclusions from these results but his ammonia is slightly raised, the glutamine is high and the citrulline slightly low.
Patient III.1 is referred at the age of 10 years and sometime after the diagnosis was made in his cousin (III.2) His mother has been concerned regarding his co-ordination from an early age and he was not walking until the age of 3. More recently there had been problems with his behaviour when he would suddenly become quite aggressive. His appetite has become less good.
At 12 months of age he had been admitted to hospital with a history of fever and sleepiness. His ammonia was found to be normal at 44 µmol/L. At that time his glutamine was not raised but his citrulline was low at 2 µmol/L. Further blood tests 2 years later showed a slightly raised blood alanine but a normal glutamine, arginine and citrulline. A urine orotic acid was only very marginally raised at 4.9 mmol/mmol of creatinine.
His health was otherwise good and he was showing normal growth. He had no particular aversion to protein containing foods. On examination there were no abnormal physical findings.
The most useful investigation will be to undertake DNA analysis in order to determine whether he has inherited the mutation from his mother.
Since his ammonia has not been found to be raised it would be inappropriate to start scavenger medication at this stage.
Commentary:
Sometimes it can be difficult to determine the pathogenesis of mutations particularly in a condition such as OTC deficiency where environmental factors such as inter-current illness, protein intake, etc may be important. For ‘mild’ OTC mutations it is not uncommon for affected individuals within the family to have very different clinical presentations or even to remain asymptomatic for many years.
In this family, decisions as to whether treatment should be started in males who are hemizygous for the mutation are difficult.
Unfortunately, this is not that unusual for the severe neonatal onset type. Reference 20 will help you understand this.
Learning objectives from this case study:
- To understand the complexity of reaching a diagnosis in individuals with less severe forms of OCT deficiency.
- To understand the role and limitations of molecular analysis in the diagnosis of OCT deficiency.
- To know the symbols used in describing family pedigrees..